Video Tutorials
Usage
Input:
- A (typically large) ligand in PDB format
- A protein receptor in PDB format
- The coordinates of the center of the binding box (i.e., the bounding box containing the binding site)
- The dimensions of the binding box, which define the size of the ligand's search space
Output:
- The three lowest-energy binding modes of the ligand as PDB files
- The representatives of the three largest clusters of ligand conformations as PDB files
- A text file containing the RMSD and energy values for the above results (dincresults.txt)
- An interactive visualization for each result using the JSMol Viewer
Sample Results
| Description | Input files | Output |
|---|---|---|
| 1OHR protein & ligand (Protease inhibitor, drug, 10 DoFs) | ligand, receptor | results page |
| 1SKJ protein & ligand (SH2 domain inhibitor, peptidomimetic, 15 DoFs) | ligand, receptor | results page |
| 4D0D protein & ligand (MHC binder, peptide, 26 DoFs) | ligand, receptor | results page |
Molecule Preparation
As part of its pre-processing steps, DINC automatically performs a number of repair and cleanup operations on the receptor and ligand provided as input.
Receptor only:
- remove water molecules
- remove chains composed entirely of non-standard amino acids
Ligand and receptor:
- remove lone pairs
- add polar hydrogens and remove non-polar hydrogens
- add Gasteiger charges
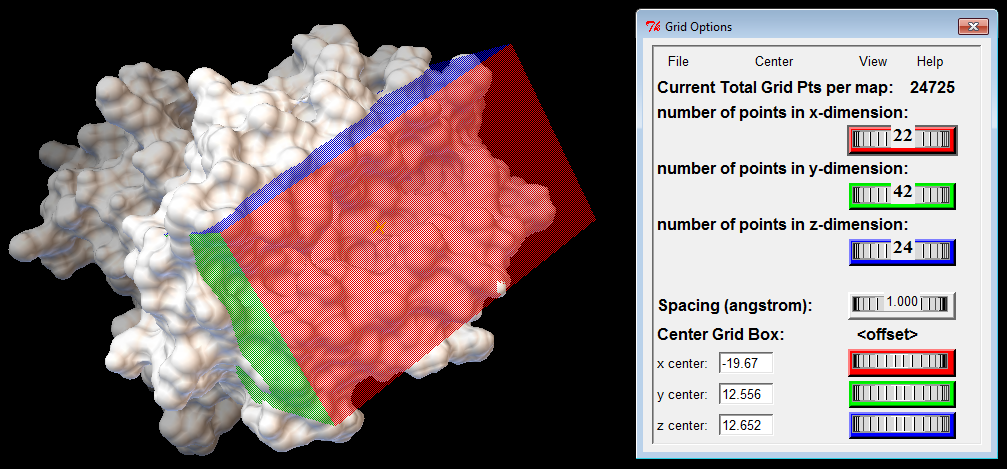
Binding Box
The binding box defines the volume that is considered for ligand sampling; it should include the receptor's binding cleft and be large enough to accommodate the ligand. The binding box also defines which atoms of the receptor are considered by the scoring function when computing binding energies. Therefore, the location and dimensions of the binding box should depend on the receptor's structure and its orientation in the input file. To find appropriate values, you should use a proper graphical interface, such as AutoDockTools (ADT), or available plugins for molecular visualization tools. Additional resources can be found at the links below.
Resources for Docking Job Preparation
- AutoDockTools
- AutoDock set-up and analysis
- AutoDock plugin for PyMOL
- AutoDock/PyMOL video tutorial
- Preparing molecules for docking with Chimera