Overview
Given the structures of a protein and ligand of interest (input), the goal of molecular docking is to predict the most likely binding mode of the resulting protein-ligand complex (output), i.e., to find the most likely conformation of this flexible ligand when it interacts with the binding site of the protein receptor. Among the main limitations of docking tools are the size and flexibility of ligands. The more flexible bonds in the ligand, the more degrees of freedom (DoFs) must be sampled, leading to a combinatorial explosion. To deal with this issue and perform molecular docking of large ligands, the Kavraki Lab at Rice University has developed DINC (Docking INCrementally). DINC uses an incremental algorithm to efficiently explore the search space of potential binding modes between a ligand and a protein. Instead of considering the entire solution space at once (i.e., docking the entire ligand), the algorithm breaks the search into several stages and optimizes each one locally before progressing to the next one.
Search Space
DINC treats the ligand as a superposition of a rigid-body component and a rotatable component. The rigid-body component contributes 6 DoFs (three for the position in space, three for the orientation), while the rotatable component contributes as many DoFs as there are rotatable bonds (or “torsions”) in the ligand. Clearly, the search space grows very large for ligands with numerous rotatable bonds. (The protein is also highly flexible and contributes so much to the complexity of the problem that docking procedures often treat it as rigid for simplicity.)
Since DINC is an incremental docking protocol, the “partial solutions” it builds correspond to overlapping fragments (i.e., contiguous sections) of the ligand. Expanding a partial solution is done by adding atoms to each fragment until the entire ligand has been reconstructed. At each stage of the algorithm, DINC considers a small number (k) of rotatable bonds as “active” in the partial solution (in addition to the 6 DoFs for position and orientation).
Algorithm
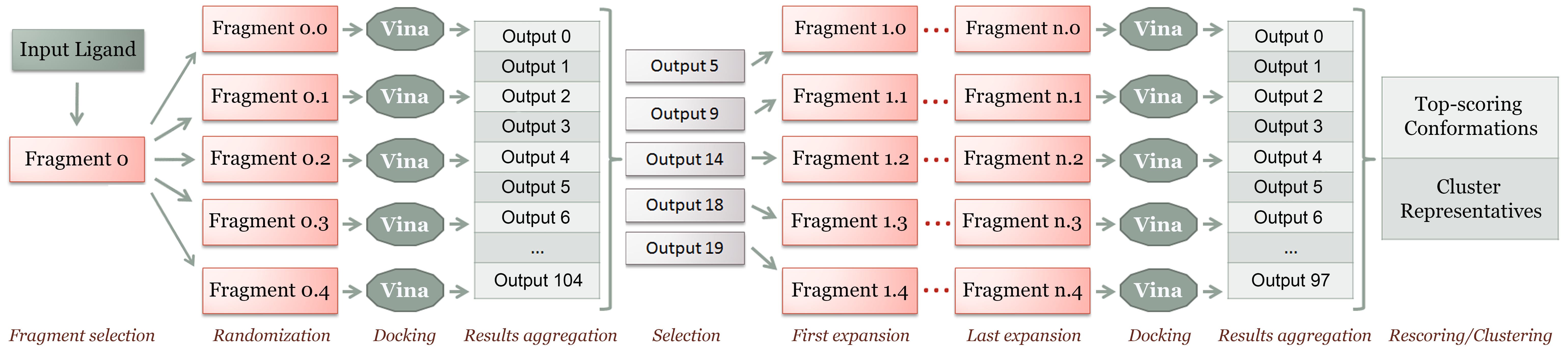
DINC is a meta-docking algorithm, in the sense that it relies on a standard docking tool, currently AutoDock Vina, to perform the sampling and scoring at each docking round. The algorithm below summarizes what was first described in (Dhanik et al., 2013). See References for details.- Choose an initial fragment containing k torsions and mark all these torsions as active.
- Randomly generate x conformations of this fragment and dock them in parallel using Vina.
- Combine the results from these independent docking tasks into a single conformation set.
- While you have not docked a fragment encompassing all atoms in the ligand, repeat:
- select the x lowest-energy conformations from the previous docking results;
- expand each fragment conformation by adding enough atoms to obtain n additional torsions;
- mark these new torsions as active; randomly select k-n torsions of the previous fragment to remain active and mark all its other torsions as rigid;
- dock all the expanded fragment conformations in parallel using Vina;
- combine the results from these independent docking tasks into a single conformation set.
- Return the lowest-energy conformations among the final docking results.