Thank you for using DINC!
You can visualize your results below. You can also download them for offline analysis.
| Ligand | 4d0d_ligand.pdb |
| Receptor | 4d0d_receptor.pdb |
| Box center | ligand center : (-11.5, -27.1, 78.0) angstrom |
| Box dimensions | ligand-based : (35.6, 22.3, 17.1) angstrom |
| Runtime | 00:26:46 |
For each ligand conformation, the binding score is reported in kcal/mol, and the heavy-atom RMSD to the original pdb conformation is reported in Å.
To move the protein-ligand complex: use 'Shift' + 'double-click' and then drag the complex to your preferred placement.
To rotate around the Z axis: hold 'Shift' and then drag horizontally.
For more things you can do within JSmol, click here.
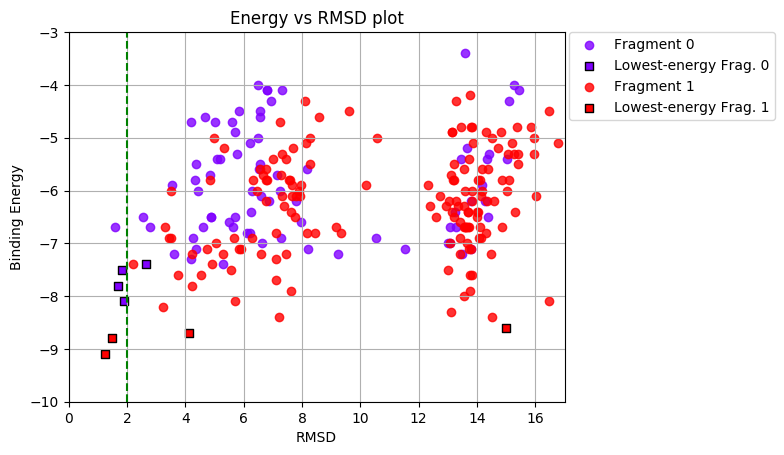
RMSD values are calculated by comparing the original pdb conformation with each fragment conformation, considering all heavy atoms of the ligand.